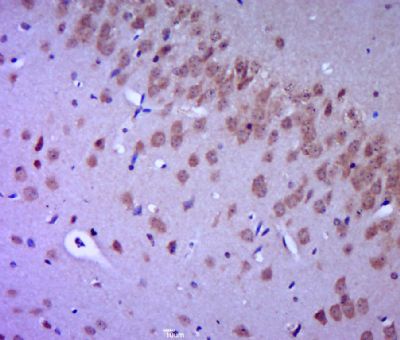
Paraformaldehyde-fixed, paraffin embedded (mouse brain tissue); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (SL0675R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining.
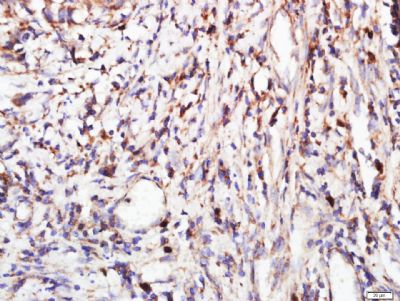
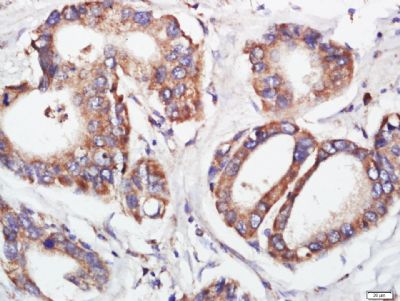
Paraformaldehyde-fixed, paraffin embedded (human stomach cancer); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (SL0675R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining.
Paraformaldehyde-fixed, paraffin embedded (human stomach cancer); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (SL0675R) at 1:400 overnight at 4°C, followed by a conjugated secondary (sp-0023) for 20 minutes and DAB staining.
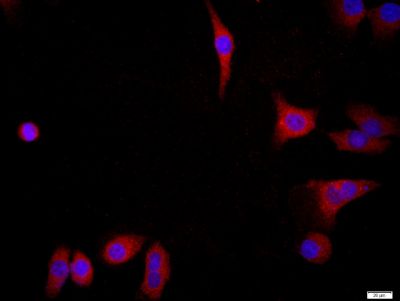
Tissue/cell: MCF7; 4% Paraformaldehyde-fixed; Triton X-100 at room temperature for 20 min; Blocking buffer (normal goat serum, SLC0005) at 37°C for 20 min; Antibody incubation with (FGFR2) Polyclonal Antibody, Unconjugated (SL0675R) 1:200, 90 minutes at 37°C; followed by a conjugated Goat Anti-Rabbit IgG antibody (SL0295G-FITC) at 37°C for 90 minutes, DAPI (blue, C02-04002) was used to stain the cell nuclei.
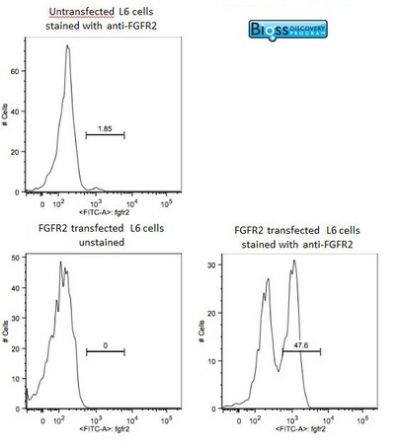
Image was kindly submitted by Dr. Uthaman from Yale University. L6 cells were transfected with FGFR2, and stained with RABBIT ANTI-FGFR2 POLYCLONAL ANTIBODY, conjugated (SL0675R-FITC) at 1:100 dilution
|