KLH conjugated synthetic peptide derived from human FGFR3:501-600/801<Cytoplasmic>
Format:
Liquid
Storage instructions:
Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles.
Concentration:
1mg/ml
Clonality:
Polyclonal
Isotype:
IgG
Applications:
WB=1:1000-2000ELISA=1:5000-10000IHC-P=1:100-500IHC-F=1:100-500IF=1:100-500(Paraffin sections need to do antigen repair)not yet tested in other applications.optimal dilutions/concentrations should be determined by the end user.
Host:
Rabbit
Product Overview:
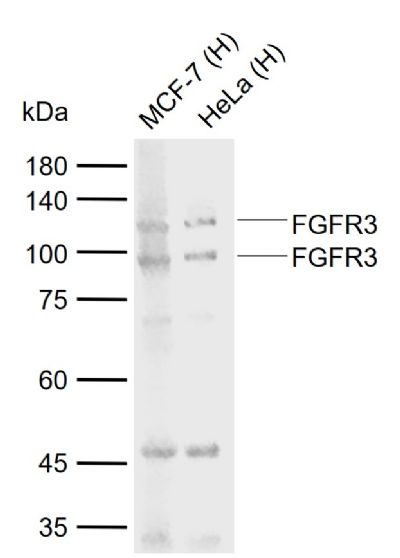
Sample: Lane 1: Human MCF-7 cell lysates Lane 2: Human HeLa cell lysates Primary: Anti-FGFR3 (SL1301R) at 1/1000 dilutionSecondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilutionPredicted band size: 86 kDaObserved band size: 100,115 kDa
This gene encodes a member of the fibroblast growth factor receptor (FGFR) family, with its amino acid sequence being highly conserved between members and among divergent species. FGFR family members differ from one another in their ligand affinities and tissue distribution. A full-length representative protein would consist of an extracellular region, composed of three immunoglobulin-like domains, a single hydrophobic membrane-spanning segment and a cytoplasmic tyrosine kinase domain. The extracellular portion of the protein interacts with fibroblast growth factors, setting in motion a cascade of downstream signals, ultimately influencing mitogenesis and differentiation. This particular family member binds acidic and basic fibroblast growth hormone and plays a role in bone development and maintenance. Mutations in this gene lead to craniosynostosis and multiple types of skeletal dysplasia. [provided by RefSeq, Aug 2017]
Function: Tyrosine-protein kinase that acts as cell-surface receptor for fibroblast growth factors and plays an essential role in the regulation of cell proliferation, differentiation and apoptosis. Plays an essential role in the regulation of chondrocyte differentiation, proliferation and apoptosis, and is required for normal skeleton development. Regulates both osteogenesis and postnatal bone mineralization by osteoblasts. Promotes apoptosis in chondrocytes, but can also promote cancer cell proliferation. Required for normal development of the inner ear. Phosphorylates PLCG1, CBL and FRS2. Ligand binding leads to the activation of several signaling cascades. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate.
Subunit: Monomer. Homodimer after ligand binding. Interacts with FGF1, FGF2, FGF4, FGF6; FGF8, FGF9, FGF10, FGF17, FGF18, FGF19, FGF20 and FGF23 (in vitro). Interacts with KLB. Affinity for fibroblast growth factors (FGFs) is increased by heparan sulfate glycosaminoglycans that function as coreceptors. Likewise, KLB increases the affinity for FGF19 and FGF21. Interacts with PIK3R1, PLCG1, SOCS1 and SOCS3.
Subcellular Location: Cell membrane; Single-pass type I membrane protein. Cytoplasmic vesicle. Endoplasmic reticulum. Note=The activated receptor is rapidly internalized and degraded. Detected in intracellular vesicles after internalization of the autophosphorylated receptor.
Tissue Specificity: Expressed in brain, kidney and testis. Very low or no expression in spleen, heart, and muscle. In 20- to 22-week old fetuses it is expressed at high level in kidney, lung, small intestine and brain, and to a lower degree in spleen, liver, and muscle. Isoform 2 is detected in epithelial cells. Isoform 1 is not detected in epithelial cells. Isoform 1 and isoform 2 are detected in fibroblastic cells.
Post-translational modifications: Autophosphorylated. Binding of FGF family members together with heparan sulfate proteoglycan or heparin promotes receptor dimerization and autophosphorylation on tyrosine residues. Autophosphorylation occurs in trans between the two FGFR molecules present in the dimer. Phosphorylation at Tyr-724 is essential for stimulation of cell proliferation and activation of PIK3R1, STAT1 and MAP kinase signaling. Phosphorylation at Tyr-760 is required for interaction with PIK3R1 and PLCG1.
Ubiquitinated. Is rapidly ubiquitinated after ligand binding and autophosphorylation, leading to receptor internalization and degradation. Subject to both proteasomal and lysosomal degradation.
N-glycosylated in the endoplasmic reticulum. The N-glycan chains undergo further maturation to an Endo H-resistant form in the Golgi apparatus.
DISEASE: Defects in FGFR3 are the cause of achondroplasia (ACH) [MIM:100800]. ACH is an autosomal dominant disease and is the most frequent form of short-limb dwarfism. It is characterized by a long, narrow trunk, short extremities, particularly in the proximal (rhizomelic) segments, a large head with frontal bossing, hypoplasia of the midface and a trident configuration of the hands.
Defects in FGFR3 are the cause of Crouzon syndrome with acanthosis nigricans (CAN) [MIM:612247]. Classic Crouzon disease which is caused by mutations in the FGFR2 gene is characterized by craniosynostosis (premature fusion of the skull sutures), and facial hypoplasia. Crouzon syndrome with acanthosis nigricans (a skin disorder characterized by pigmentation anomalies), CAN, is considered to be an independent disorder from classic Crouzon syndrome. CAN is characterized by additional more severe physical manifestation, such as Chiari malformation, hydrocephalus, and atresia or stenosis of the choanas, and is caused by a specific mutation (Ala-391 to Glu) in the transmembrane domain of FGFR3. It is proposed to have an autosomal dominant mode of inheritance.
Defects in FGFR3 are a cause of thanatophoric dysplasia type 1 (TD1) [MIM:187600]; also known as thanatophoric dwarfism or platyspondylic lethal skeletal dysplasia Sand Diego type (PLSD-SD). TD1 is the most common neonatal lethal skeletal dysplasia. Affected individuals display features similar to those seen in homozygous achondroplasia. It causes severe shortening of the limbs with macrocephaly, narrow thorax and short ribs. In the most common subtype, TD1, femur are curved.
Defects in FGFR3 are a cause of thanatophoric dysplasia type 2 (TD2) [MIM:187601]. It is a neonatal lethal skeletal dysplasia causing severe shortening of the limbs, narrow thorax and short ribs. Patients with thanatophoric dysplasia type 2 have straight femurs and cloverleaf skull.
Defects in FGFR3 are a cause of hypochondroplasia (HCH) [MIM:19200]. HCH is an autosomal dominant disease and is characterized by disproportionate short stature. It resembles achondroplasia, but with a less severe phenotype.
Defects in FGFR3 are a cause of susceptibility to bladder cancer (BLC) [MIM:22000]. A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas. They begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. Note=Somatic mutations can constitutively activate FGFR3.
Defects in FGFR3 are a cause of cervical cancer (CERCA) [MIM:603956]. A malignant neoplasm of the cervix, typically originating from a dysplastic or premalignant lesion previously present at the active squamocolumnar junction. The transformation from mild dysplastic to invasive carcinoma generally occurs slowly within several years, although the rate of this process varies widely. Carcinoma in situ is particularly known to precede invasive cervical cancer in most cases. Cervical cancer is strongly associated with infection by oncogenic types of human papillomavirus.
Similarity: Belongs to the protein kinase superfamily. Tyr protein kinase family. Fibroblast growth factor receptor subfamily. Contains 3 Ig-like C2-type (immunoglobulin-like) domains. Contains 1 protein kinase domain.
Sample:
Lane 1: Human MCF-7 cell lysates
Lane 2: Human HeLa cell lysates
Primary: Anti-FGFR3 (SL1301R) at 1/1000 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 86 kDa
Observed band size: 100,115 kDa
Product Feedback Wall
Specific References (1) | SL1301R has been referenced in 1 publications.
[IF=2.784] Ma C et al.Isolation and biological characteristic evaluation of a novel type of cartilage stem/progenitor cell derived from Small‑tailed Han sheep embryos.Int J Mol Med. 2018 Jul;42(1):525-533. ICF ; Sheep.