GTP cyclohydrolase I (also designated dopa-responsive dystonia) catalyzes the conversion of GTP to D-erythro-7,8-dihydroneopterin triphosphate, the first and rate-limiting step in tetrahydrobiopterin (SH4) biosynthesis. Tetrahydrobiopterin is an essential cofactor for 3 aromatic amino acid monooxygenases: phenylalanine, tyrosine, and tryptophan hydroxylases. Animals can synthesize tetrahydrobiopterin in vivo from GTP through several enzymatic reactions.
Function:
Positively regulates nitric oxide synthesis in umbilical vein endothelial cells (HUVECs). May be involved in dopamine synthesis. May modify pain sensitivity and persistence. Isoform GCH-1 is the functional enzyme, the potential function of the enzymatically inactive isoforms remains unknown.
Subcellular Location:
Cytoplasm. Nucleus.
Tissue Specificity:
In epidermis, expressed predominantly in basal undifferentiated keratinocytes and in some but not all melanocytes (at protein level).
Post-translational modifications:
Phosphorylated by casein kinase II at Ser-81 in HAECs during oscillatory shear stress; phosphorylation at Ser-81 results in increased enzyme activity.
DISEASE:
Defects in GCH1 are the cause of GTP cyclohydrolase 1 deficiency (GCH1D) [MIM:233910]; also known as atypical severe phenylketonuria due to GTP cyclohydrolase I deficiency;. GCH1D is one of the causes of malignant hyperphenylalaninemia due to tetrahydrobiopterin deficiency. It is also responsible for defective neurotransmission due to depletion of the neurotransmitters dopamine and serotonin. The principal symptoms include: psychomotor retardation, tonicity disorders, convulsions, drowsiness, irritability, abnormal movements, hyperthermia, hypersalivation, and difficulty swallowing. Some patients may present a phenotype of intermediate severity between severe hyperphenylalaninemia and mild dystonia type 5 (dystonia-parkinsonism with diurnal fluctuation). In this intermediate phenotype, there is marked motor delay, but no mental retardation and only minimal, if any, hyperphenylalaninemia.
Defects in GCH1 are the cause of dystonia type 5 (DYT5) [MIM:128230]; also known as progressive dystonia with diurnal fluctuation, autosomal dominant Segawa syndrome or dystonia-parkinsonism with diurnal fluctuation. DYT5 is a DOPA-responsive dystonia. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. DYT5 typically presents in childhood with walking problems due to dystonia of the lower limbs and worsening of the dystonia towards the evening. It is characterized by postural and motor disturbances showing marked diurnal fluctuation. Torsion of the trunk is unusual. Symptoms are alleviated after sleep and aggravated by fatigue and excercise. There is a favorable response to L-DOPA without side effects.
Similarity:
Belongs to the GTP cyclohydrolase I family.
SWISS:
P30793
Gene ID:
2643
Database links:
Entrez Gene: 2643 Human
Entrez Gene: 14528 Mouse
Entrez Gene: 100286831 Rabbit
Entrez Gene: 29244 Rat
Omim: 600225 Human
SwissProt: P30793 Human
SwissProt: Q05915 Mouse
SwissProt: P51596 Rabbit
SwissProt: P22288 Rat
Unigene: 62980 Human
Unigene: 86724 Human
Unigene: 10651 Mouse
Unigene: 28195 Rat
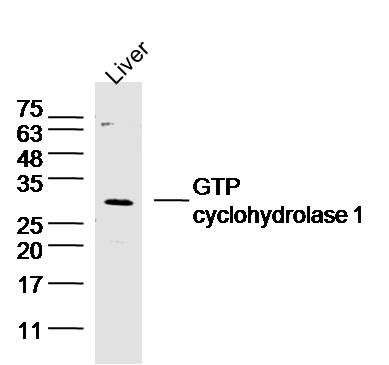
| Picture |
Sample:Liver (Mouse) Lysate at 40 ug
Primary: Anti-GTP cyclohydrolase 1 (SL0136R) at 1/300 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 27 kD
Observed band size: 27 kD
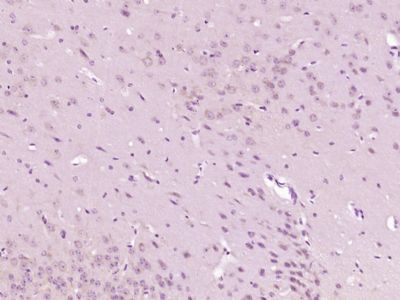
Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (GTP cyclohydrolase 1) Polyclonal Antibody, Unconjugated (SL0136R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
|
|
|