KLH conjugated synthetic peptide derived from human MeCP2:101-200/486
Format:
Liquid
Storage instructions:
Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles.
Concentration:
1mg/ml
Clonality:
Polyclonal
Isotype:
IgG
Applications:
WB=1:500-2000ELISA=1:5000-10000IHC-P=1:100-500(Paraffin sections need to do antigen repair)not yet tested in other applications.optimal dilutions/concentrations should be determined by the end user.
Host:
Rabbit
Product Overview:
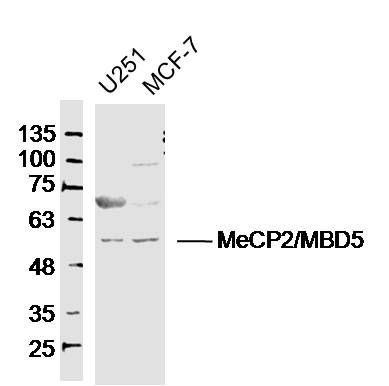
Sample:U251 (Human) Cell Lysate at 40 ugMCF-7(Human) Cell Lysate at 40 ugPrimary: Anti-MeCP2'MBD5(SL3823R)at 1/300 dilutionSecondary: IRDye800CW Goat Anti-RabbitIgG at 1/20000 dilutionPredicted band size: 52kDObserved band size: 52kD
DNA methylation is the major modification of eukaryotic genomes and plays an essential role in mammalian development. Human proteins MECP2, MBD1, MBD2, MBD3 and MBD4 comprise a family of nuclear proteins related by the presence in each of a methyl CpG binding domain (MBD). Each of these proteins, with the exception of MBD3, is capable of binding specifically to methylated DNA. MECP2, MBD1 and MBD2 can also repress transcription from methylated gene promoters. In contrast to other MBD family members, MECP2 is X-linked and subject to X inactivation. MECP2 is dispensible in stem cells, but is essential for embryonic development. MECP2 gene mutations are the cause of some cases of Rett syndrome, a progressive neurologic developmental disorder and one of the most common causes of mental retardation in females.
Function: Chromosomal protein that binds to methylated DNA. It can bind specifically to a single methyl-CpG pair. It is not influenced by sequences flanking the methyl-CpGs. Mediates transcriptional repression through interaction with histone deacetylase and the corepressor SIN3A.
Subunit: Interacts with FNBP3 (By similarity). Interacts with CDKL5
Subcellular Location: Nucleus. Note=Colocalized with methyl-CpG in the genome.
Tissue Specificity: Present in all adult somatic tissues tested.
Post-translational modifications: Phosphorylated on Ser-423 in brain upon synaptic activity, which attenuates its repressor activity and seems to regulate dendritic growth and spine maturation
DISEASE: Defects in MECP2 may be a cause of Angelman syndrome (AS) [MIM:105830]; also known as happy puppet syndrome. AS is a neurodevelopmental disorder characterized by severe mental retardation, absent speech, ataxia, sociable affect and dysmorphic facial features. AS and Rett syndrome have overlapping clinical features. Defects in MECP2 are the cause of mental retardation syndromic X-linked type 13 (MRXS13) [MIM:300055]. Mental retardation is a mental disorder characterized by significantly sub-average general intellectual functioning associated with impairments in adaptative behavior and manifested during the developmental period. MRXS13 patients manifest mental retardation associated with other variable features such as spasticity, episodes of manic depressive psychosis, increased tone and macroorchidism. Defects in MECP2 are the cause of Rett syndrome (RTT) [MIM:312750]. RTT is an X-linked dominant disease, it is a progressive neurologic developmental disorder and one of the most common causes of mental retardation in females. Patients appear to develop normally until 6 to 18 months of age, then gradually lose speech and purposeful hand movements and develop microcephaly, seizures, autism, ataxia, intermittent hyperventilation, and stereotypic hand movements. After initial regression, the condition stabilizes and patients usually survive into adulthood. Defects in MECP2 may be the cause of susceptibility autism X-linked type 3 (AUTSX3) [MIM:300496]. AUTSX3 is a pervasive developmental disorder (PDD), prototypically characterized by impairments in reciprocal social interaction and communication, restricted and stereotyped patterns of interests and activities, and the presence of developmental abnormalities by 3 years of age. Defects in MECP2 are the cause of encephalopathy neonatal severe due to MECP2 mutations (ENS-MECP2) [MIM:300673]. Note=The MECP2 gene is mutated in Rett syndrome, a severe neurodevelopmental disorder that almost always occurs in females. Although it was first thought that MECP2 mutations causing Rett syndrome were lethal in males, later reports identified a severe neonatal encephalopathy in surviving male sibs of patients with Rett syndrome. Additional reports have confirmed a severe phenotype in males with Rett syndrome-associated MECP2 mutations. Defects in MECP2 are the cause of mental retardation syndromic X-linked Lubs type (MRXSL) [MIM:300260]. Mental retardation is characterized by significantly below average general intellectual functioning associated with impairments in adaptative behavior and manifested during the developmental period. MRXSL patients manifest mental retardation associated with variable features. They include swallowing dysfunction and gastroesophageal reflux with secondary recurrent respiratory infections, hypotonia, mild myopathy and characteristic facies such as downslanting palpebral fissures, hypertelorism and a short nose with a low nasal bridge. Note=Increased dosage of MECP2 due to gene duplication appears to be responsible for the mental retardation phenotype.
Sample:
U251 (Human) Cell Lysate at 40 ug
MCF-7(Human) Cell Lysate at 40 ug
Primary: Anti-MeCP2'MBD5(SL3823R)at 1/300 dilution
Secondary: IRDye800CW Goat Anti-RabbitIgG at 1/20000 dilution
Predicted band size: 52kD
Observed band size: 52kD
Product Feedback Wall
Specific References (1) | SL3823R has been referenced in 1 publications.
[IF=2.47] Ma, Tao-tao, et al. "Geniposide alleviates inflammation by suppressing MeCP2 in mice with carbon tetrachloride-induced acute liver injury and LPS-treated THP-1 cells." International Immunopharmacology (2015). Human.