KLH conjugated synthetic peptide derived from human HMGCL:41-140/325
Format:
Liquid
Storage instructions:
Shipped at 4℃. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles.
Concentration:
1mg/ml
Clonality:
Polyclonal
Isotype:
IgG
Applications:
WB=1:500-2000ELISA=1:5000-10000IHC-P=1:100-500IHC-F=1:100-500IF=1:100-500(Paraffin sections need to do antigen repair)not yet tested in other applications.optimal dilutions/concentrations should be determined by the end user.
Host:
Rabbit
Product Overview:
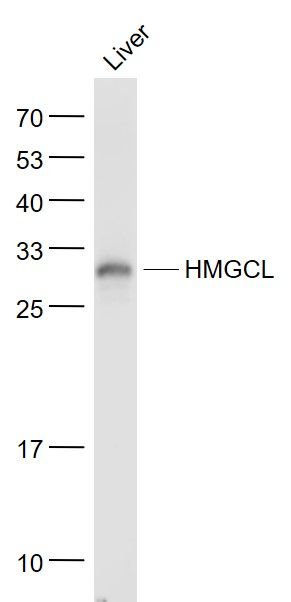
Sample: Liver (Mouse) Lysate at 40 ugPrimary: Anti- HMGCL (SL5067R) at 1/1000 dilutionSecondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilutionPredicted band size: 32 kDObserved band size: 30 kD
Hydroxymethylglutaryl-CoA lyase (HMGCL) is found in fibroblasts, liver and lymphoblasts. It has a role in ketogenesis and leucine catabolism. Defects in HMGCL are the cause of 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (hydroxymethylglutaricaciduria), an autosomal recessive disease which can lead to hypoglycemia and coma.
Function: Key enzyme in ketogenesis (ketone body formation). Terminal step in leucine catabolism.
Subunit: Homodimer; disulfide-linked. Can also form homotetramers.
Subcellular Location: Mitochondrion matrix.
Tissue Specificity: Fibroblasts, liver and lymphoblasts.
DISEASE: Defects in HMGCL are the cause of 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (HMGCLD) [MIM:246450]; also known as hydroxymethylglutaricaciduria or HL deficiency. An autosomal recessive disease affecting ketogenesis and L-leucine catabolism. The disease usually appears in the first year of life after a fasting period and its clinical acute symptoms include vomiting, seizures, metabolic acidosis, hypoketotic hypoglycemia and lethargy. These symptoms sometimes progress to coma, with fatal outcome in some cases.