6,7 dihydropteridine reductase;
DHPR;
DHPR_HUMAN;
Dihydropteridine reductase;
HDHPR;
HPR;
PKU2;
Qdpr;
Quinoid dihydropteridine reductase;
SDR33C1;
Short chain dehydrogenase/reductase family 33C, member 1.
Function:
The product of this enzyme, tetrahydrobiopterin (SH-4), is an essential cofactor for phenylalanine, tyrosine, and tryptophan hydroxylases.
Subunit:
Homodimer.
DISEASE:
Defects in QDPR are the cause of SH4-deficient hyperphenylalaninemia type C (HPASH4C) [MIM:261630]; also called dihydropteridine reductase deficiency (DHPR deficiency) or hyperphenylalaninemia tetrahydrobiopterin-deficient due to DHPR deficiency or quinoid dihydropteridine reductase deficiency (QDPR deficiency). HPASH4C is a rare autosomal recessive disorder characterized by hyperphenylalaninemia and severe neurologic symptoms (malignant hyperphenylalaninemia) including axial hypotonia and truncal hypertonia, abnormal thermogenesis, and microcephaly. These signs are attributable to depletion of the neurotransmitters dopamine and serotonin, whose syntheses are controlled by tryptophan and tyrosine hydroxylases that use SH-4 as cofactor. These patients do not respond to phenylalanine-restricted diet. HPASH4C is lethal if untreated.
Similarity:
Belongs to the short-chain dehydrogenases/reductases (SDR) family.
SWISS:
P09417
Gene ID:
5860
Database links:
Entrez Gene: 5860 Human
Entrez Gene: 110391 Mouse
Entrez Gene: 64192 Rat
Omim: 612676 Human
SwissProt: P09417 Human
SwissProt: Q8BVI4 Mouse
SwissProt: P11348 Rat
Unigene: 75438 Human
Unigene: 30204 Mouse
Unigene: 241 Rat
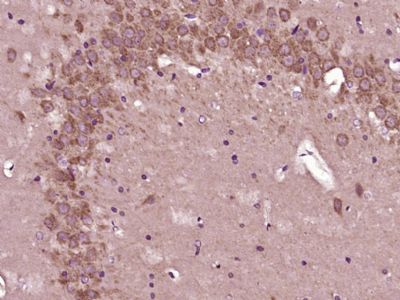
| Picture |
Paraformaldehyde-fixed, paraffin embedded (Rat brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (QDPR) Polyclonal Antibody, Unconjugated (SL7489R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.
|
|
|