The protein encoded by this gene is induced by environmental stress and developmental changes. The encoded protein is involved in stress resistance and actin organization and translocates from the cytoplasm to the nucleus upon stress induction. Defects in this gene are a cause of Charcot-Marie-Tooth disease type 2F (CMT2F) and distal hereditary motor neuropathy (dHMN). [provided by RefSeq, Oct 2008]
Function: Involved in stress resistance and actin organization.
Subunit: Interacts with TGFB1I1. Associates with alpha- and beta-tubulin, microtubules and CRYAB. Interacts with HSPB8 and HSPBAP1.
Subcellular Location: Cytoplasm. Nucleus. Cytoplasm, cytoskeleton, spindle. Note=Cytoplasmic in interphase cells. Colocalizes with mitotic spindles in mitotic cells. Translocates to the nucleus during heat shock and resides in sub-nuclear structures known as SC35 speckles or nuclear splicing speckles.
Tissue Specificity: Detected in all tissues tested: skeletal muscle, heart, aorta, large intestine, small intestine, stomach, esophagus, bladder, adrenal gland, thyroid, pancreas, testis, adipose tissue, kidney, liver, spleen, cerebral cortex, blood serum and cerebrospinal fluid. Highest levels are found in the heart and in tissues composed of striated and smooth muscle.
Post-translational modifications: Phosphorylated in MCF-7 cells on exposure to protein kinase C activators and heat shock. Phosphorylation by MAPKAPK2 and MAPKAPK3 in response to stress leads to dissociate HSP27/HSPB1 from large small heat-shock protein (sHsps) oligomers and impair its chaperone activity and ability to protect against oxidative stress effectively. Phosphorylation by MAPKAPK5 in response to PKA stimulation induces F-actin rearrangement.
DISEASE: Defects in HSPB1 are the cause of Charcot-Marie-Tooth disease type 2F (CMT2F) [MIM:606595]. CMT2F is a form of Charcot-Marie-Tooth disease, the most common inherited disorder of the peripheral nervous system. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathy or CMT1, and primary peripheral axonal neuropathy or CMT2. Neuropathies of the CMT2 group are characterized by signs of axonal regeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. Nerve conduction velocities are normal or slightly reduced. CMT2F onset is between 15 and 25 years with muscle weakness and atrophy usually beginning in feet and legs (peroneal distribution). Upper limb involvement occurs later. CMT2F inheritance is autosomal dominant. Defects in HSPB1 are a cause of distal hereditary motor neuronopathy type 2B (HMN2B) [MIM:608634]. Distal hereditary motor neuronopathies constitute a heterogeneous group of neuromuscular disorders caused by selective impairment of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs.
Similarity: Belongs to the small heat shock protein (HSP20) family.
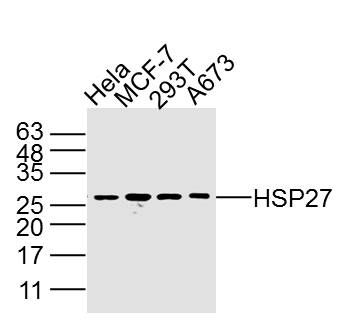
Sample:
Hela Cell (Human) Lysate at 40 ug
MCF-7 Cell (Human) Lysate at 40 ug
293T Cell (Human) Lysate at 40 ug
A673 Cell (Human) Lysate at 40 ug
Primary: Anti- HSP27 (SLM3643M) at 1/2 000 dilution
Secondary: IRDye800CW Goat Anti-Mouse IgG at 1/20000 dilution
Predicted band size: 27 kD
Observed band size: 27 kD