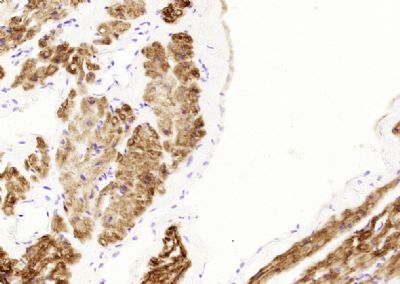
Paraformaldehyde-fixed, paraffin embedded (human heart); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (Desmin) Monoclonal Antibody, Unconjugated (SH0195) at 1:200 overnight at 4°C, followed by operating according to SP Kit(Mouse)(sp-0024) instructionsand DAB staining.